Combined Use with Other Programs
The generation of Tinker input files for structures other than peptides and nucleotides is most easily achievd through the combined use with other programs. The following example uses GaussView and MOLDEN to generate input files for geometry optimization and conformational search with Tinker.
The example chosen here explores the conformational space of n-propylcyclohexane. This system can easily be constructed from the fragments available in the GaussView builder. After construction the molecule can be save as a standard Gaussian input file such as "cyc1.com". Conversion of this file to the Tinker input file format is most easily achieved by reading in the file with molden42 cyc1.com and by writing out the structure as "cyc1.xyz" using the "Write" menu in Molden specifying the Tinker file format. At this point the input file for the system is as follows:
27 molden generated tinker .xyz (mm3 param.)
1 C 0.000000 0.000000 0.000000 1 2 6 9 10
2 C 0.000000 0.000000 1.515106 1 1 3 7 8
3 C 1.411078 0.000000 2.067037 1 2 4 17 18
4 C 2.215615 1.160661 1.517374 1 3 5 11 12
5 C 2.216276 1.160172 0.002249 1 4 6 13 14
6 C 0.805655 1.158876 -0.550551 1 1 5 15 16
7 H -0.545634 0.906562 1.887655 5 2
8 H -0.549859 -0.901910 1.890400 5 2
9 H 0.433029 -0.965385 -0.372688 5 1
10 H -1.054220 0.063502 -0.375597 5 1
11 H 1.780770 2.125272 1.889909 5 4
12 H 3.269752 1.098666 1.893397 5 4
13 H 2.765704 2.062286 -0.373071 5 5
14 H 2.762877 0.253849 -0.369606 5 5
15 H 0.299933 2.124191 -0.284547 5 6
16 H 0.840716 1.093938 -1.669099 5 6
17 H 1.916711 -0.964546 1.798419 5 3
18 C 1.364377 0.086165 3.603915 1 3 19 20 21
19 H 2.361016 0.065608 3.992744 5 18
20 H 0.886440 0.997954 3.895664 5 18
21 C 0.571131 -1.110379 4.161271 1 18 22 23 24
22 H -0.476441 -0.898305 4.111106 5 21
23 H 0.786626 -1.982818 3.580485 5 21
24 C 0.975456 -1.356491 5.626723 1 21 25 26 27
25 H 0.839215 -0.457118 6.190166 5 24
26 H 0.364882 -2.132066 6.039741 5 24
27 H 2.003197 -1.651289 5.668466 5 24
|
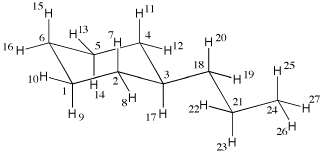 |
The first line contains the number of atoms in the input file (here 27) together with a comment (here stating that the structure has been generated with molden). The second line specifies the first center with running number "1", being of atom type "C" (a carbon atom), followed by the xyz coordinates, the first atom being located at the origin of the coordinate system in this particular case. The sixth column specifies the MM3 force field atom type (here 1) and (through a maximum of four additional n umbers) the connectivity of the current center. Center 1 is in this case connected to centers 2 and 6 (two carbon atoms) and to centers 9 and 10 (two hydrogen atoms). It is clear from this connectivity that atom 1 must be one of the ring carbon atoms in the cyclohexane ring. The other centers are defined in an analogous manner.
Before being able to explore the conformational space of the system we still need to generate an appropriate keyword file "cyc1.key" defining the force field to be used for the present study. The most appropriate force field for hydrocarbons and related organic molecules ist the MM3 force field and a minimal keyword file will accordingly be:
parameters /usr/local/tinker/params/mm3
This line points to the MM3 parameter file that can also be inspected here. The atom types 1 and 5 are described at the very beginning of the parameter file as belonging to sp3 hybridized carbon atoms as present in alkanes and as hydrogen atoms connected to carbon atoms:
atom 1 C "CSP3 ALKANE" 6 12.000 4
atom 2 C "CSP2 ALKENE" 6 12.000 3
atom 3 C "CSP2 CARBONYL" 6 12.000 3
atom 4 C "CSP ALKYNE" 6 12.000 2
atom 5 H "EXCEPT ON N,O,S" 1 1.008 1
Searching the conformational space can again be achieved with the scan program from the Tinker program suite. An appropriate command file named "cyc1.run" including some sorting and cleanup is as follows:
scan cyc1 0 5 100 0.0001
grep "Map" cyc1.log | cat > tempo
sort tempo -nr -k 6,6n -o cyc1.list
rm tempo
After making the file executable this command file can be executed with:
./cyc1.run >& cyc1.log &
Conformational search with the MM3 force field gives in this particular case rise to 7 different conformers with the following MM3 force field energies (in kcal/mol):
Potential Surface Map Minimum 1 12.2568
Potential Surface Map Minimum 3 12.9967
Potential Surface Map Minimum 6 13.2390
Potential Surface Map Minimum 4 14.8350
Potential Surface Map Minimum 7 15.1229
Potential Surface Map Minimum 2 15.8632
Potential Surface Map Minimum 5 15.9882
The structures of these conformational minima are located in a series of files called "cyc1.001" . . . "cyc1.007". A combined coordinate file can be generated from these single structure files using the
archive
program of the Tinker program suite, which works quite selfexplanatory. Using the UNIX command "cat" it is also possible to combine the single structure files into one archive, now also reflecting the sequence of relative stability:
cat cyc1.001 cyc1.003 cyc1.006 cyc1.004 cyc1.007 cyc1.002 cyc1.005 > cyc1.arc
The single structure files as well as the archive files generated with archive or the cat command can be read with Molden and converted from there again to input files for quantum mechanical calculations with Gaussian. On analyzing the results obtained for n-propylcyclohexane you will realize that the conformers differ in the orientation of the n-propyl side chain only. This is due to the conformational search algorithm used in scan.