Construction and Optimization of Peptides
Building up a peptide is achieved using the protein subprogram in either an interactive mode (when the program will prompt you for input) or by putting the parameters into an input file and calling the program together with the input file through a shell script. A list of amino acids and their abbreviations can be found here. The input file (termed here testz1.dat) for building up the dialanine tripeptide with N-terminal acetyl and C-terminal N-Me amide capping groups is as follows:
| testz1 ACE-(ala)2-NME ACE ala ala NME n |
 |
The first of these lines specifies the output file (here: testz1), the second line a title in free format (here: ACE-(ala)2-NME), the third line the N-terminal residue, here taken to be a capping acetyl group (ACE), the fourth and fifth lines specify the two alanine residues without any additional parameters, and the sixth line the C-terminal N-methyl amide capping group (NME). One empty line terminates the definition of the amino acid sequence. After specifying a non-cyclic peptide (n) the definition of the peptide is finished. The input file described above can be executed simply from the command line:
protein < testz1.dat
The above example assumes a matching parameter file (testz1.key) to exist containing additional information such as the force field type and parameters to use. In case a matching keyword file does not (yet) exist, the protein subprogram prompts the user for the corresponding information. In case we want to use the amber99 parameters for the AMBER force field, the testz1.key file is quite simple:
parameters /usr/local/tinker/params/amber99
This file is also needed in order to read the newly generated testz1.xyz file into FFE.
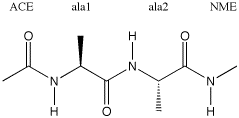
The example chosen above generates a peptide structure in which all phi angles are set to -135.0 degrees, all psi angles to +135.0 degrees, and all omega angles to 180.0 degrees. This generates an approximately linear peptide chain found as substructures in beta sheets. The definition of these angles is shown below for the ala-ala dipeptide, carrying an acetyl protecting group at the N-terminal end, and capping the C-terminal end as N-methyl amide.
The value of omega (the dihedral angle describing rotation around the peptide bond) is often very close to 180.0 degrees (a trans-peptide bond). In some selected cases, however, a cis-peptide bond (with values for omega around 0.0 degrees) can also be found in protein structures. To which of the two amino acids participating in a peptide bond the omega angle is attributed is a matter of personal choice. In the Tinker input file the omega angle given for amino acid i defines the peptide bond between amino acid i and i+1. The values of phi and psi depend on the particular folding motive of the protein and the following typical cases are often observed: (a) a right-handed helical turn (alpha helix) with phi = -55.0 and psi = -45.0 degrees; (b) beta sheet structures are formed by extended chains with phi = -135.0 and psi = +135.0 degrees (approximate values).
The angle information can be included in the protein input file on the same line as the residue itself. The following example shows the construction of the end-capped alanine decapeptide in a conformation with phi and psi angles equal to -60.0 degrees, generating a rough starting conformation for a partial alpha helix. Due to the presence of the two capping groups, omega angles can be defined for both the leading and the tailing alanine residue:
testz1
testz1 ACE-(ala)10-NME, alpha helix input
ACE
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
NME
n
The xyz-coordinate files generated with protein can be minimzed with a number of different optimization routines. Optimization of the peptide structure to a loose convergence criterion of around 0.01 kcal/mol/angstrom can be achieved with the minimize program, which is also known to work well with larger protein structures. As input the program requires the principal name of the system (here: testz1) and the convergence criterion:
minimize testz1 0.01
The output consists of a new xyz-coordinate file as well as some information concerning the convergence of the optimization process. In order to direct the latter into a log file, the construction of the peptide and its optimization with minimize can be combined in one shell script (e.g. testz1.run):
protein < testz1.dat
minimize testz1 0.01
This short script can be executed (after making it executable with "chmod u+x testz1.run") with the following command:
./testz1.run > testz1.log
The alanine decapeptide system can also be used to illustrate the performance of the various optimizers such as minimize, newton, and optimize contained in Tinker. The table below shows the overall CPU times, cycle numbers, and final energy of the optimized structure for various optimizers for convergence criteria of 0.01 and 0.001 kcal/mol/angstrom, respectively:
| optimizer | timea,c (sec) |
cyclesa | final energya (kcal/mol) |
timeb,c (sec) |
cyclesb | final energyb (kcal/mol) |
|
|---|---|---|---|---|---|---|---|
| minimize | 13.79 | 1099 | -18.9569 | 15.52 | 1255 | -18.9574 | |
| newton | 4.62 | 31 | -18.6278 | 5.18 | 32 | -18.6278 | |
| optimize | 34.38 | 1499 | -10.6792 | 35.03 | 1521 | -10.6792 |
aconvergence criterion of 0.01 kcal/mol/angstroms; bconvergence criterion of
0.001 kcal/mol/angstroms; call times have been obtained using the UNIX time
command
We can see, that for small systems such as the one chosen here, the newton optimizer is much more efficient, the difference to the minimize optimizer being increasingly large with increasing convergence criteria. It can also be seen that the newton optimizer performs a small number of expensive steps, while the minimize optimizer uses a very larger number of cheap steps. While these two optimizers converge onto essentially the same final structure, this is not the case with the optimize optimizer. The latter is not only the slowest optimizer, but also converges onto a much less favorable structure. These findings are, however, only relevant for small systems such as the alanine decapeptide chosen here.