Searching Conformational Space
In order to search the conformational space of larger systems, Tinker offers a number of different subprograms. The strategies that can be used for the exploration of conformational space can roughly be divided in those relying on (a) systematic searching conformational space or on (b) random search strategies.
One of the options to search the conformational space of macromolecules systematically is the combination of large torsional motion with local geometry optimization as implemented in the program scan. The first argument accepted by the program is the principal file name (which helps locate the xyz-coordinates file used for structural input). The second argument accepted by the program decides over the selection of torsional angles in the search list. Automatic selection is chosen with the argument 0. Other options for the second argument are 1 (selecting manual selection of angles to rotate) or 2 (manual selection of angles to freeze). In case automatic selection of dihedral angles is specified, the number of torsions is determined by the program and listed in the output file. For each conformational minimum found in the search list, the program searches along a given number of normal modes for new minima by taking a larger step in the direction (positive as well as negative) along one normal mode, followed by geometry optimization to the next local minimum. The number of search directions represents the third argument accepted by the program (default = 5). The list of conformational minima maintained by the program encompasses structures within a given energy window. The size of this window can be given (in kcal/mol) as the fourth argument to the program. The window size defaults to 100 kcal/mol. The local geometry optimization to a minimum energy structure depends on a convergence criterion in the same way as already discussed for the different optimizers. The convergence criterion is the fifth argument given to the program and defaults to 0.0001 kcal/mol/angstrom. While a tigher convergence criterion will slow down the conformational search, a looser convergence criterion will make it more difficult to match identical conformations obtained in two different local searches. The following example builds the end-capped amino acid alanine (often termed the alanine dipeptide) through an appropriate call to the protein program (building up the peptide structure in file testz1.xyz) and searches the conformational spcace of this system with scan. A shell script executing all of these steps (e.g. saved in file testz1.run) is as follows:
protein < testz1.dat
scan testz1 0 5 100 0.0001
grep "Map" testz1.log | cat > tempo
sort tempo -nr -k 6,6n -o testz1.list
rm tempo
This script file can be executed (after making it executable) in a straigh forward manner with:
./testz1.run >& testz1.log &
The input file for protein is as follows:
| testz1 testz1 ACE-ala-NME, alanine dipeptide ACE ala NME n |
 |
The keyword file for this example is also quite simple:
parameters /usr/local/tinker/params/amber99
enforce-chirality
The keyword enforce-chirality checks that during conformational searches the absolute configuration of the system is maintained. The output generated by scan is as follows:
Normal Mode Local Search Minimum 4
Search Direction 1 Step 38 -17.2591
Search Direction 2 Step 8 -20.4180
Search Direction 3 Step 22 -16.0053
Potential Surface Map Minimum 10 -16.0053
.
.
This part of the output file reflects the investigation of structures generated from minimum 4 by searching along the first three search directions. The first two searches yield structures which are known already. The third search, converged after 22 geometry optimization steps, yields a new conformational minimum, which at this point happens to be number 10 in the list of known conformational minima.
After execution of the program scan the working directory contains one new structure file (in xyz coordinates) for each conformation found during the conformational search, named testz1.001, testz1.002 . . . What the remaining lines in the command file testz1.run do is analyze the output file testz1.log in order to find the energetically most favorable structure. This is achieved here by extracting all lines containing the string "Map" from output file testz1.log, writing these lines to the file tempo, then using the UNIX sort program to sort the lines in file tempo in reverse numerical order (-nr) using the values in field number 6 (-k 6,6n), and writing the sorted list to a new file testz1.list. Finally, the file tempo ist deleted. For the current example scan finds 28 distinct conformational minima. The fourth minimum found during the search with an energy of -22.65 kcal/mol turns out to be the most favorable one. The structure of this conformation as generated by ffe is shown at the left side below.
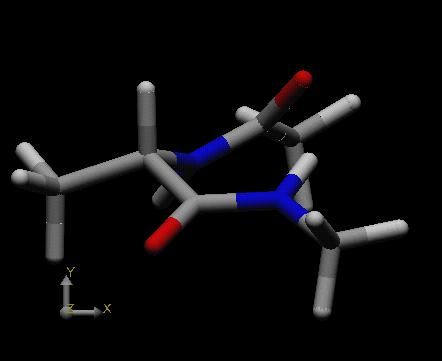 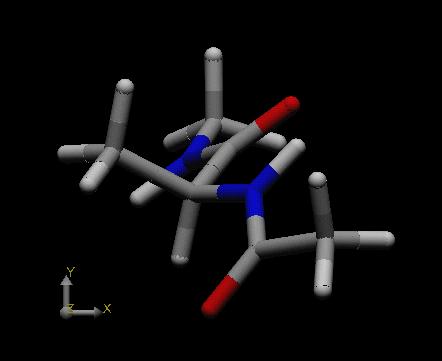 |
This conformation contains a hydrogen bond between the acetyl protecting group of the alanine nitrogen atom and the N-H bond of the N-Me capping group of the alanine carboxylate terminus. Since an overall seven-membered ring is formed (including the hydrogen atom) and the alanine methyl group is positioned in a pseudo-equatorial position of the seven-membered ring, this conformation is referred to as the "C7eq" conformation of the dipeptide. The second most favorable structure is the "C5" conformation shown at the right side above with an energy of -21.75 kcal/mol.
Even for moderately large systems an exhaustive search of conformational space quickly becomes prohibitive. In order to limit the conformational searches performed with scan, it is therefore often quite reasonable, to limit the search to selected dihedral angles. Using the end-capped hexaalanine heptapeptide as an example, an approximate structure for the alpha-helical conformation can be generated easily with the following input file for protein:
test1
test1 ACE-(ala)6-NME, alpha helix input
ACE
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
ala -60.0 -60.0 180.0
NME
n
Optimization of this structure with the newton optimizer can be performed in a straight forward manner as discussed before, ultimately yielding (using the AMBER force field and the amber99 parameter set) an optimized local minimum with an energy of -17.6165 kcal/mol. The conformational space of this system can be explored to a limited degree involving only the essential degrees of freedom of the N- and C-terminal amino acid residues using the following input for scan:
test1
1
7 8
8 9
57 58
58 59
4
100.0
0.0001
In this particular case the input file contains the principal name of the system (here: test1), the manual selection of dihedral angle parameters (1), and the definition of four bonds around which rotation will be allowed (7/8, 8/9, 57/58, 58/59). Inspection of the molecular structure of this system with ffe shows, that these dihedral angles are the phi and psi angles of the two terminal alanine residues in the system. After terminating the input of rotatable bonds with one blank line, the number of dihedral angles to explore for each conformer found during the search is set to 4, the energy window for all conformers is set to 100.0 kcal/mol, and the geometry optimization convergence criterion is set to 0.0001 kcal/mol/angstrom. This limited conformational search proceeds in a rather short time to find 21 different conformers, the best of which has an energy of -17.6931 kcal/mol. The seed structure obtained from initial geometry optimization with newton is found as the second best structure. Please observe that the length of conformational searches also depends on the dihedral angles chosen as the active parameters in the input file. The selection of dihedral angles in the middle of the peptide sequence is, for example, an effective tool for searching a much larger conformational space than selection of dihedral angles located at the end of the peptide sequence.
When studying large, conformationally flexible systems, one is often primarily interested in the energetically most favorable structure, the "global minimum". One method for searching for the global minimum energy structure is the global search trajectory method "Sniffer" by Butler and Slaminka. The method is available in the Tinker suite in the subprogram sniffer. The algorithm used in sniffer attempts to locate the global minimum of the system through a combination of molecular dynamics steps and gradient optimization steps. As input the program accepts the principal file name of the systems (which helps to locate the xyz-coordinate input file), the number of geometry perturbation steps to generate initial sets of trial structures (defaulting to 100 steps), the target energy of the global minimum (defaulting to 0.0 kcal/mol), the RMS optimization criterion for local geometry optimization (defaulting to 1.0 kcal/mol/angstrom). Should the energy of the system fall below what is defined as target energy, the conformational search will be terminated (regardless of the value of the current gradient). This requires the user to play with the target energy in order to find meaningful lower bounds.
The success of sniffer in locating the global minimum also depends on the input structure used. This can be demonstrated using again the alanine dipeptide example used before. Generating this system with all psi, phi, omega angles set to 180.0 degrees (linear peptide chain) with the following input file to protein:
| testz1 testz1 ACE-ala-NME ACE ala 180.0 180.0 180.0 NME n |
|
and handing the resulting xyz-coordinates file testz1.xyz to sniffer with:
sniffer testz1 100 -25.0 1.0
locates the C5 conformation of the alanine dipeptide at -21.74 kcal/mol within 2471 iterations as the supposedly global minimum. From the conformational search performed with scan it is clear, that this is indeed one of the energetically most favorable conformations of this system, but not the global minimum. This is not so much a consequence of the number of trial structures (10, 100, 200, or 500 trial structures yielding the same final result), but the linear peptide conformation used as the starting structure. A non-linear peptide chain can be generated with:
testz1
testz1 ACE-ala-NME, gauche
ACE
ala 180.0 60.0 180.0
NME
n
and processed with sniffer as before. This time the C7eq minimum at -22.64 kcal/mol is indeed located within some 2154 iterations as the global minimum. While this sensitivity of the algorithm on the initial trial structure is somewhat unfavorable, the true strength of the sniffer algorithm is its speed. The last (successfull) run to locate the global minimum takes only 1.56 CPU sec., while the full conformational search with scan requires 37.60 sec. This difference is, of course, bound to increase with increasing system size.