Double-Hybrid DFT Methods
Double-hybrid density functionals (DHDFT) such as B2-PLYP (sometimes also called B2PLYP) expand the DFT exchange-correlation energies into four terms:
EXC = (1-cHF)EX(GGA) + cHFEX(HF)
+ (1-cMP2)EC(GGA) + cMP2EC(MP2) [1]
The first two terms describe the exchange energy as a mix of terms derived from GGA functionals and exact exchange (here termed EX(HF)). The actual mixing ratio of the two terms is described by the coefficient "cHF", whose value can range from 0 to 1. In a similar spirit the correlation energy is a sum of terms derived from GGA functionals and the correlation energy EC(MP2) (sometimes also called E2) calculated with second-order perturbation theory (MP2). It is important to recognize that all four terms are derived from the same Kohn-Sham orbitals.
The first DHDFT method of general applicability B2-PLYP was proposed by S. Grimme[1,2] and builds on the same exchange and correlation energy functionals used in the B3LYP hybrid functional:
EXC(B2-PLYP) = 0.47 EX(Becke) + 0.53 EX(HF)
+ 0.73 EC(Lee-Yang-Parr) + 0.27 EC(MP2) [2]
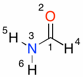
Single point B2-PLYP calculations can be performed in Gaussian 09 using the following input (here for the example of formamide):
#P blyp/cc-pVTZ iop(3/76=0470005300,3/78=0730007300) scf=tight
int=finegrid extraoverlay
8/10=90/1; 9/16=-3/6;
B2PLYP/cc-pVTZ//B3LYP/6-31G(d) SP formamide, Cs sym.
0 1
C -0.1620723564 0.3875637772 0.
O -1.200221649 -0.2454918984 0.
N 1.0860148709 -0.1581628487 0.
H -0.1292709682 1.4957821901 0.
H 1.1841617934 -1.1644188162 0.
H 1.9167313093 0.414367596 0.
|
 |
This instructs the program to run through a first step, in which the first three terms of energy expression [2] are computed. This can also be seen in the output file from the settings in link 301, where a scaling factor of exact exchange is specified with ScaHFX= 0.530000, the scaling factor for (local and GGA- corrected) exchange energies is given by ScaDFX= 0.470000 0.470000, and the scaling factor for DFT correlation energies is given by 0.730000 0.730000 :
IExCor= 402 DFT=T Ex=B+HF Corr=LYP ExCW=0 ScaHFX= 0.530000
ScaDFX= 0.470000 0.470000 0.730000 0.730000 ScalE2= 1.000000 1.000000
The energy reported after completion of the SCF as:
SCF Done: E(RB+HF-LYP) = -169.638324821 A.U. after 11 cycles
represents the first three terms in energy expression [2]. The program then continues with calculation of the E2 correlation energy in links 801 and 906:
Range of M.O.s used for correlation: 1 132
.
.
E2 = -0.7813197650D+00 EUMP2 = -0.17041964458627D+03
Scaling the correlation energy of -0.7813197650D+00 Hartree reported in the output file with 0.27 and addition to the SCF energy of -169.638324821 Hartree then yields the final B2-PLYP energy of -169.84928116 Hartree. The above sequence also illustrates that, in terms of computational effort, B2-PLYP calculations are as expensive as MP2(FULL) calculations on Hartree-Fock orbitals and thus much more expensive (on larger molecular systems) as compared to hybrid DFT methods such as B3LYP.
More recent versions of Gaussian implement gradients and second derivatives for B2-PLYP and facilitate its use through a "B2PLYP" keyword. For the sake of computational efficiency, this latter option defaults to the frozen core approximation for the PT2 calculation. Reproducing the B2-PLYP energy calculated with manual scaling of the E2 correlation energy described above thus requires an explicit statement of a "FULL" electron count in the PT2 calculations:
#P B2PLYP(FULL)/cc-pVTZ scf=tight int=finegrid
B2PLYP/cc-pVTZ//B3LYP/6-31G(d) SP formamide, Cs sym., all electrons correlated
0 1
C -0.1620723564 0.3875637772 0.
O -1.200221649 -0.2454918984 0.
N 1.0860148709 -0.1581628487 0.
H -0.1292709682 1.4957821901 0.
H 1.1841617934 -1.1644188162 0.
H 1.9167313093 0.414367596 0.
The program output will now report all scaling factors in energy expression [2] in link 301:
IExCor= 419 DFT=T Ex+Corr=B2PLYP ExCW=0 ScaHFX= 0.530000
ScaDFX= 0.470000 0.470000 0.730000 0.730000 ScalE2= 0.270000 0.270000
and the full B2-PLYP energy at the end of link 906 as:
Range of M.O.s used for correlation: 1 132
.
.
E2(B2PLYP) = -0.2109563352D+00 E(B2PLYP) = -0.16984928115650D+03
As mentioned above the "B2PLYP" keyword defaults to the frozen-core approximation and the resulting B2-PLYP energy will therefore be somewhat smaller as compared to the original version. The input file:
#P B2PLYP/cc-pVTZ scf=tight int=finegrid
B2PLYP/cc-pVTZ//B3LYP/6-31G(d) SP formamide, Cs sym., FC approx.
0 1
C -0.1620723564 0.3875637772 0.
O -1.200221649 -0.2454918984 0.
N 1.0860148709 -0.1581628487 0.
H -0.1292709682 1.4957821901 0.
H 1.1841617934 -1.1644188162 0.
H 1.9167313093 0.414367596 0.
|
|
thus yields:
Range of M.O.s used for correlation: 4 132
.
.
E2(B2PLYP) = -0.1984755522D+00 E(B2PLYP) = -0.16983680037351D+03
The same energy can be obtained "by hand" with a small modification of the original input file mentioned above:
#P blyp/cc-pVTZ iop(3/76=0470005300,3/78=0730007300) scf=tight
int=finegrid extraoverlay
8/10=4/1; 9/16=0/6;
B2PLYP/cc-pVTZ//B3LYP/6-31G(d) SP formamide, Cs sym., FC approx.
0 1
C -0.1620723564 0.3875637772 0.
O -1.200221649 -0.2454918984 0.
N 1.0860148709 -0.1581628487 0.
H -0.1292709682 1.4957821901 0.
H 1.1841617934 -1.1644188162 0.
H 1.9167313093 0.414367596 0.
which yields:
SCF Done: E(RB+HF-LYP) = -169.638324821 A.U. after 13 cycles
and
E2 = -0.7350946427D+00 EUMP2 = -0.17037341946401D+03
and, after scaling E2 with 0.27 and summing up, a B2-PLYP energy of -169.836800375 Hartree. One may argue that the consideration of all electrons in the PT2 calculation is required simply because the calculation of GGA correlation energies is also based on the full electron density. However, the time savings derived from the FC approximation are significant and one may thus opt for this variant in all studies of larger molecular systems. Whether B2-PLYP calculations are run with the "FC" or "FULL" approximation is not necessarily obvious from the corresponding program output and may also be responsible for different B2-PLYP energies obtained from different programs or program versions.
The choice of optimum mixing coefficients in double hybrid DFT methods depends significantly on the chosen reference data set. A B2-PLYP modification optimized for the description of kinetic data termed B2K-PLYP was developed by J. M. L. Martin and coworkers[3] and uses the following expression for the exchange-correlation energies:
EXC(B2K-PLYP) = 0.28 EX(Becke) + 0.72 EX(HF)
+ 0.58 EC(Lee-Yang-Parr) + 0.42 EC(MP2) [3]
In a similar spirit the variant B2T-PLYP has been optimized for the description of thermochemical data,[3] while B2GP-PLYP is intended as a General Purpose functional.[4]
Furthermore, the choice of optimum mixing coefficients depends significantly on the chosen basis set. This has been explored by L. Radom and coworkers[4] in the context of optimizing B2-PLYP variants for open shell species. Restricted-open shell (or ROB2-PLYP) variants were found to perform slightly better as compared to unrestricted versions in their description of thermochemical data of open-shell species. Whether restricted or unrestricted open-shell calculations are performed is specified by modifications of the "B2PLYP" keyword, here demonstrated for the aminyl radical in its 2B1 state as an example:
#P ROB2PLYP/cc-pVTZ scf=tight int=finegrid
ROB2PLYP/cc-pVTZ//ub3lyp/6-31G(d), NH2 radical, C2v sym.
0 2
H 0.000000 0.804274 -0.506307
H 0.000000 -0.804274 -0.506307
N 0.000000 0.000000 0.144659
|
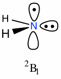 |
The respective unrestricted calculation can be chosen using the "UB2PLYP" keyword. Using the rather large cc-pVQZ basis set for parameter optimization, the best performance was found for the following ROB2-PLYP variant:
EXC(ROB2-PLYP(59,28)) = 0.41 EX(Becke) + 0.59 EX(HF)
+ 0.72 EC(Lee-Yang-Parr) + 0.28 EC(MP2) [4]
However, when comparing the performance of this variant with that of the original B2-PLYP parameterization using the more economical cc-pVTZ (or other polarized triple-zeta basis sets), the difference is rather small. This changes again for small double-zeta basis sets such as cc-pVDZ, where the new parameterization enjoys significant advantages.
Literature:
1) S. Grimme,
"Semiempirical hybrid density functional with perturbative second-order correlation"
J. Chem. Phys. 2006, 124, 034108-16.
2) F. Neese, T. Schwabe, S. Grimme,
"Analytic derivatives for perturbatively corrected “double hybrid” density functionals: Theory, implementation, and applications"
J. Chem. Phys. 2006, 126, 124115-15.
3) A. Tarnopolsky, A. Karton, R. Sertchook, D. Vuzman, J. M. L. Martin,
"Double-Hybrid Functionals for Thermochemical Kinetics"
J. Phys. Chem. A 2008, 112, 3-8.
4) A. Karton, A. Tarnopolsky, J.-F. Lamere, G. C. Schatz, J. M. L. Martin,
"Highly Accurate First-Principles Benchmark Data Sets for the Parametrization and Validation of Density Functional and Other Approximate Methods. Derivation of a Robust, Generally Applicable, Double-Hybrid Functional for Thermochemistry and Thermochemical Kinetics"
J. Phys. Chem. A 2008, 112, 12868–12886.
5) D. C. Graham, A. S. Menon, L. Goerigk, S. Grimme, L. Radom,
"Optimization and Basis-Set Dependence of a Restricted-Open-Shell Form of B2-PLYP Double-Hybrid Density Functional Theory"
J. Phys. Chem. A 2009, 113, 9861-9873.
last changes: 23.10.2014, HZ questions & comments to: zipse@cup.uni-muenchen.de