8.4. Influence of Theoretical Methods
The results of chemical shielding calculations depend on the chosen quantum mechanical methods, basis sets, and solvation models (if any). Best results are obtained when using basis sets specifically optimized for the accurate description of electron densities close to the artomic nucleus. A typical example is the "pcSseg-n" family of basis sets optimized by F. Jensen for the accurate description of shielding values, where index n ranges from 0 to 4.[1,2] The basis set family has been optimized for use with hybrid DFT methods, but works quite well also with correlated methods such as the RI-MP2 implementation available in ORCA. The smallest practically useful member of the family is pcSseg-1, which is of "double zeta plus polarization (DZP)" quality in terms of the number of basis functions available for valence space orbitals and thus similar in design as the Pople-style 6-31G(d,p)[3-5] or the Ahlrichs def2-SVP basis set.[6] The next larger family members pcSseg-2 and pcSseg-3 are then of "triple zeta (TZP)" and "quadrupe zeta (QZP)" quality, respectively. QZP-quality basis sets are actually quite large already and chemical shielding calculations with even larger basis sets become very demanding even for small molecular systems. Basis sets such as those of the "pcSseg-n" family have been optimized for shielding calculations by (among others) addition of high-exponent (tight) s- and/or p-type basis functions, whose exponents have been optimized such as to minimize the errors in shielding calculations for a reference data set. We will test the performance of these basis sets in predicting the 13C NMR shieldings and chemical shifts of 1,4-dimethylbenzene (para-xylene). We will restrict ourselves here to the C2/C3/C5/C6 positions, which are expected to have identical shieldings/shifts due to the Ci symmetry of the most stable conformer of this system. The isotropic shieldings obtained in RI-MP2/<basis> calculations are summarized in Figure 8.4.1a. For the systematically designed pcSseg-n basis sets we see a systematic decrease of shieldings with increasing basis set size, the difference between the values for the smallest (pcSseg-1) and the largest (pcSseg-3) basis sets amounting to 65.74 - 60.78 = 4.96 ppm. A similar trend can also be observed in the shieldings calculated with the general purpose Ahlrichs def2-nVP basis sets, but the differences between the smallest (def2-SVP) and largest (def2-QZVPP) basis sets are now much larger at 83.55 - 63.19 = 20.35 ppm, and particularly the results for the small def2-SVP basis set seem surprisingly poor. This is also the case fore the (general purpose) Pople-style 6-31G(d,p) and 6-311G(2df,2pd) basis sets. In summary we see from the results for 1,4-dimethylbenzene in Figure 8.4.1a that (i) spezifically optimized basis sets such pcSseg-n show a much more systematic basis set size dependence of shielding values as compared to general purpose basis sets, and that (ii) the differences in shielding values calculated with optimized or general-purpose basis sets are particularly large for the smallest (DZP quality) membes of these basis set series.
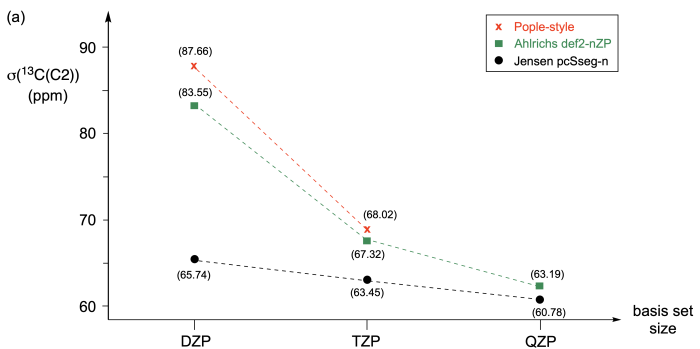
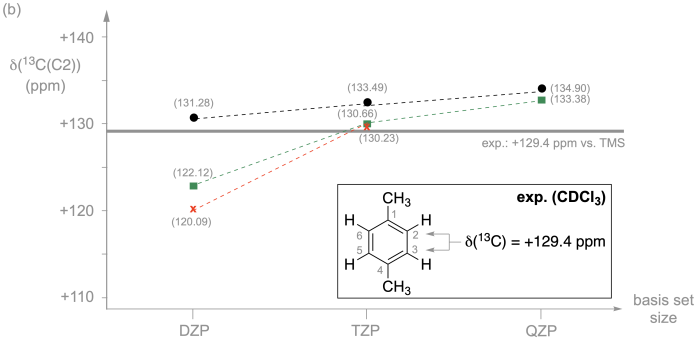
Figure 8.4.1.(a) 13C Isotropic shielding values calculated for the C2/C3/C5/C6 positions of 1,4-dimethylbenzene at RI-MP2/<basis> level of theory with ORCA. Results for the Pople-style 6-31G(d,p) and 6-311G(2df,2pd) basis sets are shown in red, for the Ahlrichs def2-SVP, def2-TZVPP and def2-QZVPP basis sets in green, and the Jensen pcSseg-1, pcSseg-2, and pcSseg-3 basis sets in black. (b) 13C Chemical shift values calculated for the C2/C3/C5/C6 positions of 1,4-dimethylbenzene at RI-MP2/<basis> level of theory with ORCA using TMS as the reference. The experimentally measured 13C chemical shift for these four carbon atoms in CDCl3 amounts to +129.4 ppm vs. TMS.
How the calculation of 13C chemical shift values at the C2 position of 1,4-dimethylbenzene calculated relative to TMS as the reference is impacted by basis set choice is shown in Figure 8.4.1b. For the pcSseg-n basis sets we see a systematic trend towards larger (more positive) shift values with increasing basis set size. The shift value predicted with the pcSseg-3 basis set amounts to +134.90 ppm, which is 5.5 ppm higher than the experimental value of +129.4 ppm (in CDCl3). Similar trends can also be identified for the def2-nVP and the Pople-style basis sets, the largest differences in predicted chemical shifts appearing for the smallest (DZP quality) members of the basis set series.
What type of quantum mechanical methods are best suited for NMR chemical shift predictions depends, in part dramatically, on the systems of interest. For main group elements recent benchmark studies suggest that dispersion-corrected double hybrid DFT methods such as D3-DSD-PBEP86 provide the highest accuracy.[7] But we still have to keep in mind that we currently perform gas phase (and not solution phase) calculations with quantum mechanical methods considered to be very good (but not outstanding) for NMR shielding calculations.
References
[1] F. Jensen, "Basis Set Convergence of Nuclear Magnetic Shielding Constants Calculated by Density Functional Methods", J. Chem. Theory Comp. 2008, 4, 719−727.
[2] F. Jensen, "Segmented Contracted Basis Sets Optimized for Nuclear Magnetic Shielding", J. Chem. Theory Comp. 2015, 11, 132−138.
[3] P. C. Hariharan, J. A. Pople, "The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies", Theor. Chim. Acta 1973, 28, 213−222.
[4] R. Krishnan, J. S. Binkley, R. Seeger, J. A. Pople, "Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions", J. Chem. Phys. 1980, 72, 650−654.
[5] M. J. Frisch, J. A. Pople, J. S. Binkley, "Self-consistent molecular orbital methods. 25. Supplementary functions for Gaussian basis sets", J. Chem. Phys. 1984, 80, 3265−3269.
[6] F. Weigend, R. Ahlrichs, "Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy", Phys. Chem. Chem. Phys. 2005, 7, 3297−3305.
[7] C. J. Schattenberg, M. Kaupp, "Extended Benchmark Set of Main-Group Nuclear Shielding Constants and NMR Chemical Shifts and Its Use to Evaluate Modern DFT Methods", J. Chem. Theory Comput. 2021, 17, 7602−7621.
last changes: 12.12.2024, Imon Mandal, and Hendrik Zipse, supported by the RTG 2620 on "Ion Pair Effects in Molecular Reactivity" questions & comments to: zipse@cup.uni-muenchen.de