8.5. Selecting Appropriate Reference Systems
The 13C NMR results presented for 1,4-dimethylbenzene in the previous chapter involve tetramethylsilane (TMS) not only as the zero of the 1H, 13C, and 29Si NMR scales, but also as the reference system relative to which we compare the computed shielding values. Looking at these two compounds from a chemical point of view, we note that TMS is not neccesarily the best choice as a reference because (i) we compare C(sp2) hybridized carbon atoms in the 1,4-dimethylbenzene substrate with C(sp3) hybridized carbon atoms in the TMS reference, and (ii) we compare substrate and reference 13C atoms with a comparatively large chemical shift difference of 129.4 ppm. That both factors are of some importance in accurate 13C chemical shift predictions can be illustrated by switching from TMS to benzene (C6H6) as the reference molecule and using eq. (4) rather than eq. (5) for converting calculated shieldings to chemical shifts. When measured in CDCl3, benzene displays a single signal in the proton-decoupled 13C NMR spectrum at +128.37 ppm. The chemical shifts of the C2 carbon atom in 1,4-dimethylbenzene and the carbon atoms in benzene thus differ by only 1.5 ppm, and both centers are of C(sp2) type. That this leads to a complete change in the predictive ability of 13C NMR chemical shift predictions can be seen graphically in Figure 8.5.1, where we employ the same levels of theory as already in Figure 8.4.1. The vertical axis for 13C NMR chemical shifts has been rescaled in this case to accomodate the fact that ALL predictions for the C2 center in 1,4-dimethylbenzene fall into a range of only one ppm. We also add the commonly assumed error for 13C NMR measurements of ±0.5 ppm as a faint gray bar just to indicate, that all computationally predicted 13C NMR chemical shifts fall into the experimental error range. While we may still feel most comfortable when using the largest possible and specifically optimized basis sets for NMR shielding calculations, the results in Figure 8.5.1 actually illustrate that highly accurate NMR shift predictions are possible even with DZP-quality basis sets when appropriate reference systems are selected.
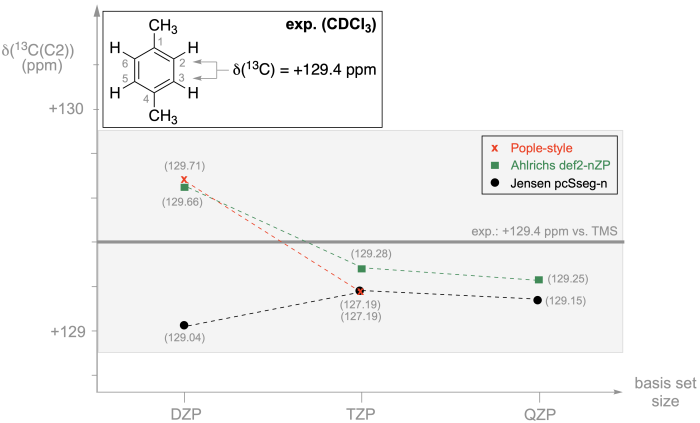
Figure 8.5.1. 13C Chemical shift values calculated for the C2/C3/C5/C6 positions of 1,4-dimethylbenzene at RI-MP2/<basis> level of theory with ORCA using benzene (C6H6) as the reference. The experimentally measured 13C chemical shift for the C2 position in 1,4-dimethylbenzene is +129.4 ppm and that for benzene is +128.37 ppm in CDCl3.
The selection of "appropriate" reference systems becomes more important in 31P NMR shift calculations, where the zero of the NMR scale is defined by 85% H3PO4 in water. What the molecular identity of this reference system is at this (high) concentration is not easily seen, and selection of water as a medium additionally complicates comparison to, for example, organophosphorus compounds in less polar solvents. One can, of course, simply ignore the apparent complexity and use phosphoric acid as the reference system in 31P shielding calculations. The results obtained for triphenylphosphane (PPh3, a frequently used reagent and catalyst in organic synthesis) and its oxide are shown in Figure 8.5.2. In CDCl3 solution PPh3 displays a signal at -4.7 ppm in the proton-decoupled 31P NMR spectrum, often accompanied by a small amount of its oxide (OPPh3) with a signal at +29.5 ppm.[1] Gas phase shielding calculations at RI-MP2/pcSseg-2//M06-2X/def2-TZVPP level of theory performed with ORCA yield the results shown in Figure 8.5.2.
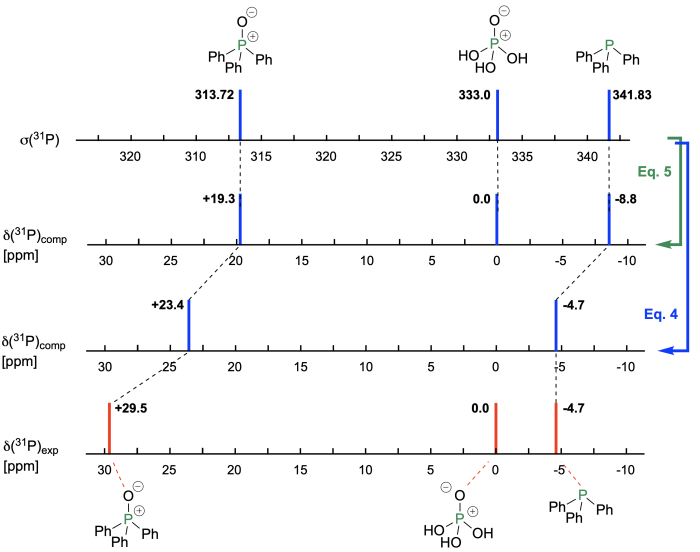
Figure 8.5.2. 31P Chemical shift values calculated for the phosphorus atom in PPh3 and OPPh3 relative to phosphoric acid as the reference system.
Application of eq. (5) predicts -8.8 ppm for PPh3 and +19.3 ppm for its oxide OPPh3. The significant offset for both signals towards more negative/less positive shift values may either indicate substantial differential solvent effects between the gas phase calculations, the water medium for phosphoric acid, and CDCl3 for PPh3 and its oxide. Or may also indicate that monomeric phosphoric acid is not a sufficiently good model for the 85% H3PO4 reference system. We can separate these two effects by selecting PPh3 as the reference system with a known chemical shift value in CDCl3 solution of -4.7 ppm, and applying eq. (4) for the prediciton of the chemical shift value for OPPh3. As shown in Figure 8.5.2 this reduces the error for the shift prediction of OPPh3 by close to 4 ppm. The predictive value of NMR chemical shift calculations can thus be enhanced very significantly by selecting a small set of reference compounds with closely similar chemical and spectroscopic properties as the system under investigation.[2]
In order to make this strategy less dependent on the accuracy of a SINGLE analytical measurement, it is common place to select a small set of reference compounds with similar, but not necessarily identical spectroscopic properties (a focused library). This idea can be further extended by linear regression analysis of computed chemical shieldings for a larger dataset with the respective experimentally measured shift data. The slope and offset parameters of these correlations can then be used for the prediction of NMR chemical shifts of new compounds (or for the validation of NMR shift assignments of known compounds).[3] The combination of shielding calculations with statistical analysis protocols has largely aided the 1H and 13C NMR assignments of conformationally flexible compounds in organic chemistry.[4]
References
[1] C. Lindner, Y. Liu, K. Karaghiosoff, B. Maryasin, H. Zipse, "The Aza-Morita–Baylis–Hillman Reaction: A Mechanistic and Kinetic Study, Chem. Eur. J. 2013, 19, 6429−6434.
[2] J. Helberg, Y. OE, H. Zipse, "Mechanistic Analysis and Characterization of Intermediates in the Phosphane-Catalyzed Oligomerization of Isocyanates", Chem. Eur. J. 2018, 24, 14387–14391.
[3] M. W. Lodewyk, M. R. Siebert, D. J. Tantillo, "Computational Prediction of 1H and 13C Chemical Shifts: A Useful Tool for Natural Product, Mechanistic, and Synthetic Organic Chemistry", Chem. Rev. 2012, 112, 1839–1862.
[4] N. Grimblat, M. M. Zanardi, A. M. Sarotti, "Beyond DP4: an Improved Probability for the Stereochemical Assignment of Isomeric Compounds using Quantum Chemical Calculations of NMR Shifts", J. Org. Chem. 2015, 80, 12526–12534.
last changes: 12.12.2024, Imon Mandal, and Hendrik Zipse, supported by the RTG 2620 on "Ion Pair Effects in Molecular Reactivity" questions & comments to: zipse@cup.uni-muenchen.de