Visualization of Molecular Orbitals using MOLDEN
MOLDEN can be used to visualize molecular orbitals in two ways:
- by reading in all required information from the Gaussian output file
- by reading in a cube file containing information for only one orbital
1) Reading in all required information from the Gaussian output file
- run a single point calculation at the desired level of theory including the pop=full and gfinput keywords. Only then will MOLDEN find all required information in the output file. For the formaldehyde example chosen before this can be achieved with the following input:
#P HF/STO-3G scf=tight pop=full gfinput HF/STO-3G//HF/STO-3G sp formaldehyde 0 1 C1 O2 1 r2 H3 1 r3 2 a3 H4 1 r3 2 a3 3 180.0 r2=1.21672286 r3=1.10137241 a3=122.73666566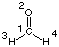
Remember that the output files can become quite sizeable with this output option.
- start MOLDEN and read in the output file using the Read button
- choose Density Mode from the control panel. MOLDEN will then switch to a different perspective showing a 2D-projection of the molecule on a regular grid.
- select the molecular orbital to be visualized through the Orbital button. The appearing popup-window lists the orbitals from top to bottom with increasing orbital energies and also provides the occupancy of the orbitals in a third column. The HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) can be selected directly using the corresponding buttons.
- In order to obtain a space filling 3D-presentation of the molecular orbitals, the Space option must be selected from the control panel. After providing a cutoff value for the electron density contour, a 3D-plot of the orbital will be constructed in an iterative procedure. Meaningful values for the cutoff value are in the range of 0.02 - 0.2 for occupied orbitals in the valence shell, but might be significantly different for diffuse unoccupied or very compact core orbitals. In order to select a different cutoff value, simply reselect the Space button. How different the appearance of molecular orbitals can be depending on the cutoff value is illustrated in the following plots showing the HOMO of formaldehyde (HF/STO-3G level) at cutoff values of 0.02, 0.04, 0.08, and 0.2:
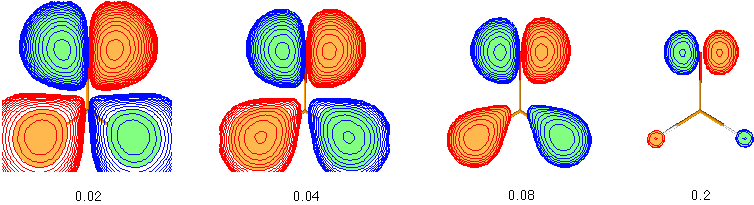
It can clearly be seen that the more diffuse parts of the molecular orbitals centered around the hydrogen atoms in the lower part of the pictures is dominating at low cutoff values such as 0.02, but that the more compact upper part describing an oxygen centered lone pair becomes more dominating at higher cutoff values such as 0.2. This illustrates that the question of "orbital shape" is difficult to answer even in a qualitative way. - The total molecular electron density (the sum over all occupied molecular orbitals) can be viewed using the Density button. The cutoff value used for these plots can again be set using the Space button.
- the Fill button toggles between a space filling and a mesh-based presentation.
- in some cases, it is more instructive to analyze a cut through the molecular orbitals. This option is supported by the Euclid button. The plotting plane used in this case can be chosen using the PlotPlane menu.
2) Reading in a cube file containing information for only one orbital
The values for a given molecular orbital at regularly spaced points in space can be computed in all three dimensions producing a grid of density values. This grid can be computed in an automated manner with the cube keyword. Together with sensible defaults concerning the grid size and origin this provides a convenient way of producing plots for single molecular orbitals. Please observe that cube files can become quite large.
- run a single point calculation at the desired level of theory including the cube=orbitals keyword. In addition to the actual keyword some information must be included in the input file in order to specify the cube file name as well as the number and type of orbitals. If only one number is given, only information on this orbital is dumped to the cube file. The following input file will produce a cube file named mycubefile_HOMO.cub containing a 3D-representation of molecular orbital 11 (the HOMO) of ally cation calculated at the HF/6-31G(d) level of theory.
#P HF/6-31G(d) cube=orbitals scf=(tight) HF/6-31G(d) HOMO allyl cation + generation of cube-file +1 1 H,0,0.,0.,-1.5682867937 C,0,0.,0.,-0.4948409695 C,0,1.1776939788,0.,0.2112484936 C,0,-1.1776939788,0.,0.2112484936 H,0,2.1316522244,0.,-0.2859258635 H,0,1.1894473911,0.,1.2871012074 H,0,-2.1316522244,0.,-0.2859258635 H,0,-1.1894473911,0.,1.2871012074 mycubefile_HOMO.cub 11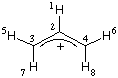
- after starting MOLDEN, switch to the Density Mode and read in the cube file using the Read Cube button at the bottom of the control panel. Please observe that MOLDEN will only read cube files containing information for a single orbital.
More recent versions of Gaussian do not fully support the cube keyword anymore. It is then advisable to generate the cubes separately from the formatted checkpoint file using the cubegen utility program. How this can be combined with generation of the formatted checkpoint file is shown below for the example of the allyl cation at the HF/6-31G(d) level using the full PBS job script:
#!/bin/csh
#PBS -l mem=128mb
#PBS -q long
setenv g03root /usr/local
setenv GAUSS_SCRDIR /scratch
setenv GAUSS_EXEDIR /usr/local/g03b3
setenv GAUSS_ARCHDIR /usr/local/g03b3
setenv LD_LIBRARY_PATH "${GAUSS_EXEDIR}:/usr/lib"
cat >$GAUSS_SCRDIR/$PBS_JOBNAME << EOF
%chk=/scratch/allyl.chk
%mem=6000000
#P HF/6-31G(d) scf=(tight) formcheck
HF/6-31G(d) HOMO allyl cation + generation of cube-file
+1 1
H,0,0.,0.,-1.5682867937
C,0,0.,0.,-0.4948409695
C,0,1.1776939788,0.,0.2112484936
C,0,-1.1776939788,0.,0.2112484936
H,0,2.1316522244,0.,-0.2859258635
H,0,1.1894473911,0.,1.2871012074
H,0,-2.1316522244,0.,-0.2859258635
H,0,-1.1894473911,0.,1.2871012074
EOF
touch $PBS_O_WORKDIR/$PBS_JOBNAME.$HOST
/usr/local/g03b3/g03 < $GAUSS_SCRDIR/$PBS_JOBNAME > $GAUSS_SCRDIR/$PBS_JOBNAME.log
mv $GAUSS_SCRDIR/$PBS_JOBNAME.log $PBS_O_WORKDIR/$PBS_JOBNAME.log
mv $GAUSS_SCRDIR/$PBS_JOBNAME.chk $PBS_O_WORKDIR/$PBS_JOBNAME.chk
rm -f $GAUSS_SCRDIR/$PBS_JOBNAME
mv Test.FChk $PBS_O_WORKDIR/$PBS_JOBNAME.fch
$GAUSS_EXEDIR/cubegen 0 MO=11 $PBS_O_WORKDIR/$PBS_JOBNAME.fch $PBS_O_WORKDIR/$PBS_JOBNAME.cub 0 h
exit
What the additional lines after execution of Gaussian do is move the Gaussian output files from /scratch to the users working directory, move the formatted checkpoint file named "Test.FChk" to a more meaningful name (here: allyl.fch), and then call cubegen to generate the cube file for molecular orbital 11. This latter program accepts six arguments on the command line. The first argument (here: 0) defines the memory allocated to the run. Using the argument of 0 specifies automatic memory allocation. The second argument defines the type of cube plot desired in this run. The particular argument here (MO=11) defines the molecular orbital 11, other numbers indicating other orbitals. The third argument defines the name of the formatted checkpoint file, the fourth argument the name of the cube file, the fifth argument the number of points per side of the cube. Using the argument of 0 at this point again leads to automatic selection of sensible defaults. Finally, the last argument indicates, whether a header (h) is to be included in the cube file. If this is not desired, the last argument must be n.
The above job script assumes that the name of the script itself is "allyl". For other names to work, the name of the checkpoint file must be altered.